Pólipo colorretal é qualquer estrutura que faz saliência na luz intestinal. São tumores benignos, parecidos com verrugas, que se desenvolvem na mucosa (revestimento interno da parede) do cólon e reto. A maior preocupação é a capacidade que alguns pólipos têm de evoluir e se transformar em câncer.
Introdução e Sintomas.
Diagnóstico dos pólipos colorretais.
Classificação Macroscópica e Microscópica (histológica).
Sequência Adenoma-Carcinoma e Serrilhado-Carcinoma.
Carcinoma “de novo”.
Síndromes hereditárias associadas ao câncer colorretal.
Polipose Adenomatosa Familiar ou FAP
Câncer Colorretal Hereditário Sem Polipose (HNPCC).
Outras Poliposes Colônicas.
Tratamento dos Pólipos.
Estudo dos pólipos pela colonoscopia.
Técnica da retirada dos pólipos por colonoscopia.
Complicações da polipectomia e mucosectomia.
Pólipo com Câncer Colorretal (maligno).
Seguimento após a retirada de pólipo colorretal (seguimento pós-polipectomia).
Quando parar com a colonsocopia de prevenção?
Recomendações em casos especiais.
Quando antecipar a colonoscopia seguimento.
Pólipo colorretal é qualquer estrutura que faz saliência na luz intestinal. São tumores benignos, parecidos com verrugas, que se desenvolvem na mucosa (revestimento interno da parede) do cólon e reto. A maior preocupação é a capacidade que alguns pólipos têm de evoluir e se transformar em câncer.
Geralmente os pólipos colorretais são assintomáticos, mas podem:
1. Sangrar, saindo pelo ânus sangue vivo (hematoquezia) ou escuro (melena) podendo causar anemia por deficiência de ferro;
2. Produzir muco (catarro), saindo pelo ânus isoladamente ou misturado ao sangue e quando abundante pode causar desidratação com diminuição do potássio;
3. Quando grandes podem obstruir parcialmente (causa dor e inchaço abdominal) ou totalmente (abdome agudo obstrutivo) o intestino e/ou causar alternância do hábito intestinal (ora diarreia ora constipação).
A descoberta e retirada dos pólipos evita o câncer colorretal, mas a presença não é para pânico:
1. Os pólipos são comuns, e estão presentes em cerca de 30% dos adultos com 45 anos ou mais;
2. Nem todos os tipos de pólipo têm a capacidade de evoluir para o câncer de intestino;
3. São necessários alguns anos (10 a 15 anos dependendo do tamanho e da classificação) para que o pólipo se transforme em câncer, já que o mesmo é um tumor benigno;
4. A maioria dos pólipos pode ser retirada com segurança pela colonoscopia.
O acompanhamento médico das pessoas que apresentaram pólipos dependerá do número, tamanho, localização e tipo das lesões (classificação de risco).
Os pólipos acometem homens e mulheres de todas as raças, mas são mais frequentes nas pessoas que vivem em centros industrializados, o que mostra uma clara associação entre o estilo de vida e dieta.
Em relação ao estilo de vida, os pólipos são mais comuns em obesos, pessoas com dietas ricas em carne vermelha e gorduras e pobres em fibras, e em fumantes.
A idade também é um fator importante para o surgimento dos pólipos, visto que mais de 90% dos cânceres de intestino acontecem após os 50 anos (idade em que se inicia a prevenção). Os pólipos surgem 5 a 10 anos antes.
Outros fatores de risco é a história familiar e a genética, por isso deve ser dada atenção para a presença de pólipos e câncer colorretal (de intestino) em parentes próximos, principalmente jovens, e quando mais de 2 membros da mesma família foram acometidos (independe da idade).
Como regra geral, a pesquisa de pólipos se inicia mais cedo em pessoas com história familiar de pólipos ou câncer colorretal.
Diagnostico dos pólipos colorretais
Através da colonoscopia diagnóstica, isto é, quando existe indicação clínica como na hemorragia digestiva baixa, dor abdominal associado alteração do hábito intestinal, anemia, diarreia crônica, etc. Nesse caso o pólipo pode ser a causa da indicação ou apenas um achado acidental.
Através da colonoscopia de prevenção em pacientes assintomáticos.
RISCO MÉDIO – VEJA DETALHES ➡ ⇒
Idade igual ou maior a 50 anos. Recentemente, a American Cancer Society (ACS) publicou novas diretrizes para abordagem da doença e anunciou que o rastreamento do câncer colorretal deve começar aos 45 anos para adultos com risco médio. Veja a revisão aqui! ➡
RISCO INTERMEDIÁRIO – VEJA DETALHES ➡ ⇒
História familiar de câncer colorretal, adenoma e pólipo serrilhado em seus parentes de primeiro grau (pais, filhos e irmãos).
RISCO ALTO – VEJA DETALHES ➡ ➡ ⇒
A- Colonoscopia anterior com diagnóstico de pólipo (adenoma ou pólipo serrilhado) no intestino.⇒
B- Quem já teve câncer colorretal. ➡ ⇒
C- Mulheres com câncer de mama, ovário ou útero. ➡ ⇒
D- Portadores da Doença de Crohn e Retocolite Ulcerativa. ➡ ⇒
Os pólipos do cólon são classificados de acordo com a sua característica macroscópica e microscópica.
Classificação Macroscópica
Classificação de Paris
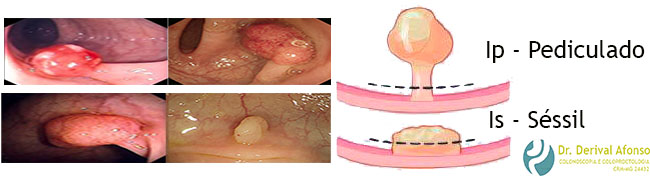 Ip – lesão pediculada
Ip – lesão pediculada
Is – lesão séssil
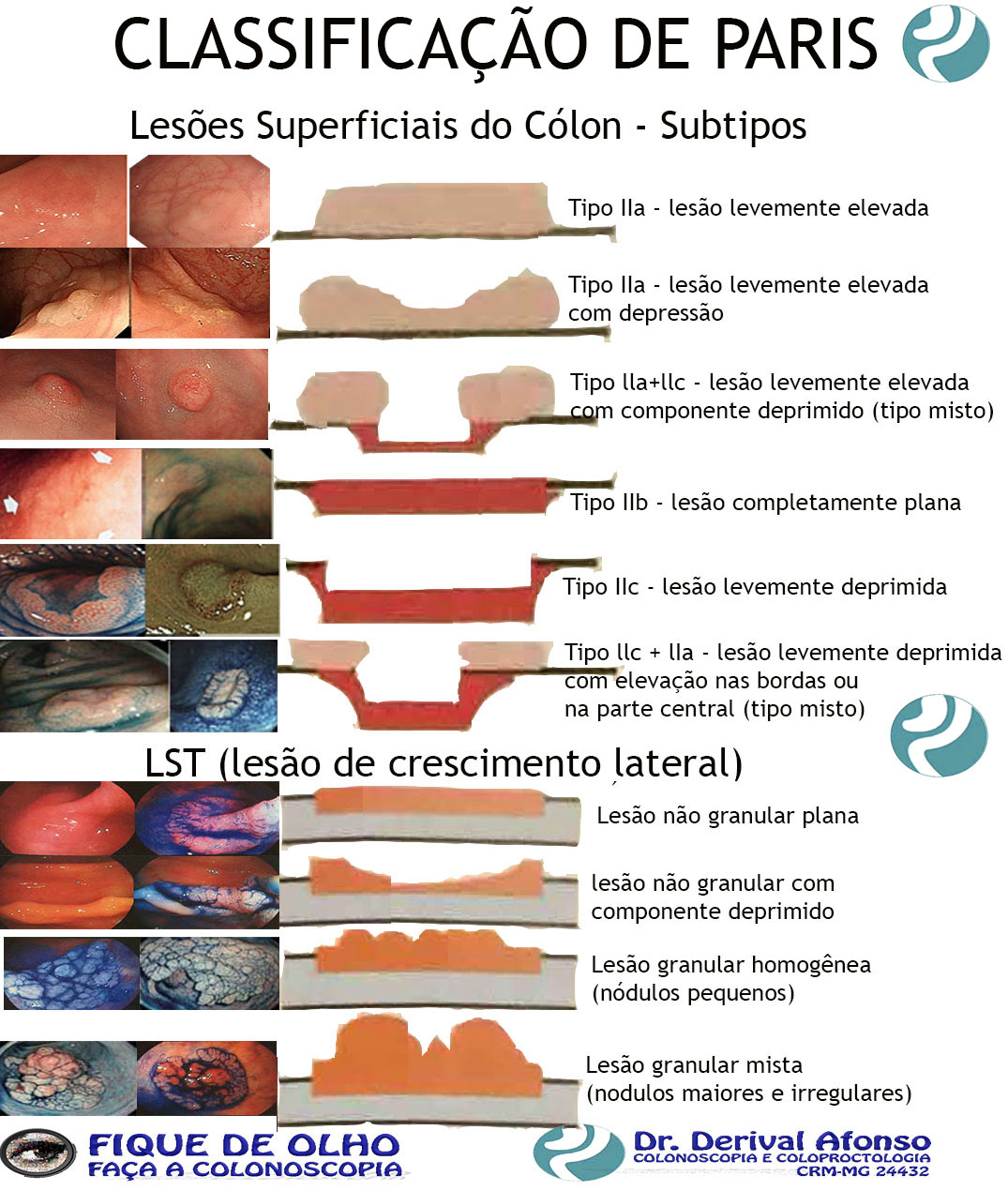 Lesões Superficiais do Cólon
Lesões Superficiais do Cólon
ᴥ» Tipo IIa – lesão levemente elevada;
ᴥ» Tipo IIb – lesão completamente plana;
ᴥ» Tipo IIc – lesão levemente deprimida;
ᴥ» Tipo III – lesão ulcerada;
ᴥ» Tipo lla+llc – lesão levemente elevada com componente deprimido (tipo misto);
ᴥ» Tipo llc + lIa – lesão levemente deprimida com elevação nas bordas ou na parte central (tipo misto). Lesões planas elevadas (IIa) e lesões sésseis (Is) podem ser confundidas. A diferenciação se faz medindo a altura da lesão com pinça de biópsia fechada, quando for maior que 2,5 mm é considerada séssil.
LST (lesão de crescimento lateral)
Considerada quando a lesão superficialmente elevada é maior que 10 mm. São classificadas em:
ᴥ» Lesão granular homogênea (nódulos pequenos);
ᴥ» Lesão granular mista (nodulações maiores e irregulares);
ᴥ» Lesão não granular plana;
ᴥ» Lesão não granular com componente deprimido.
Classificação Microscópica (histológica)
Não Neoplásicos
1. Hiperplásicos
2. Inflamatórios
3. Pseudopólipos
4. Hamartomas
Neoplásicos
1. Adenoma
a. Adenoma tubular.
—b. Adenoma tubulo-viloso.
—c. Adenoma viloso.
2. Serrilhado
—a. Adenoma serrilhado séssil
—b. Adenoma serrilhado séssil tradicional
—c. Misto
Não Neoplásicos – Hiperplásicos
São os mais comuns, 30% a 40 % de todos os pólipos do cólon e reto. Formados pela hiperproliferação das células normais da mucosa. São geralmente pequenos (menores que 5 mm), pálidos ou brilhantes, localizados na porção mais distal (final) do intestino e reto e que quase sempre desaparecem com a distensão do reto.
 ᴥ» Os pólipos hiperplásicos puros menores que 10 mm não têm potencial de se tornarem câncer e não necessitam de seguimento após a retirada.
ᴥ» Os pólipos hiperplásicos puros menores que 10 mm não têm potencial de se tornarem câncer e não necessitam de seguimento após a retirada.
ᴥ» Os pólipos hiperplásicos maiores que 10 mm tem um comportamento semelhante aos pólipos serrilhados com risco de virar câncer e necessitam de seguimento após a retirada.
A Polipose Hiperplásica foi descrita em 1980 (Williams GT. Histopathology 1980; 4: 155–70). Deve-se pensar na possibilidade dessa polipose quando múltiplos, grandes e/ou proximais ao retossigmóide. Diferem dos pólipos hiperplásicos isolados, pois podem atingir até 3 cm. Apesar de afetar preferencialmente pacientes entre a sexta e sétima décadas, existem casos descritos em jovens. A polipose hiperplásica é considerada uma condição pré-maligna e, portanto pode evoluir para o câncer colorretal.
 Os critérios para o diagnóstico da polipose hiperplásica são:
Os critérios para o diagnóstico da polipose hiperplásica são:
1. Pelo menos cinco pólipos hiperplásicos diagnosticados histopatologicamente proximais ao cólon sigmóide, dos quais dois sejam maiores que 10 mm ou;
2. Qualquer número de pólipos hiperplásicos proximais ao cólon sigmóide em um indivíduo com um parente de primeiro grau com polipose hiperplásica ou;
3. Mais que 30 pólipos hiperplásicos de qualquer tamanho distribuídos pelo cólon.
Inflamatórios ![]()
São lesões resultantes do tecido de granulação em regeneração. Geralmente únicos e podem atingir tamanhos consideráveis e tornarem-se pediculados quando existe risco de sangramento e obstrução intestinal. Pode ter uma aparência endoscópica sinistra, irregular, avermelhado e ulcerado ou apenas regular e brancacento. Não apresentam risco de transformação maligna e não necessitam de seguimento pós-polipectomia. Quando maiores de 10 mm devem ser removidos por serem semelhantes e endoscopicamente indistinguíveis dos pólipos neoplásicos.
Pseudopólipos ![]()
São ilhas de mucosa residual após cicatrização de processos inflamatórios e ulcerados no cólon e reto de longa duração. Geralmente são múltiplos de várias formas. Não apresentam risco de transformação maligna e não necessitam de ressecção e muito menos de seguimento pós-polipectomia.
Hamartomas 
São coleções de tecidos normais (compõem a lâmina própria) organizadas de modo anormal e desorganizada.
Os pólipos juvenis são hamartomas comumente encontrados em crianças abaixo de 10 anos, mas podem aparecer também em adolescentes e adultos, sendo sua incidência maior no sexo masculino. São cistos de retenção e criptas revestidas por células normais. Pode ulcerar e conter infiltrado inflamatório. O pedículo caracteristicamente é fino sem elemento muscular, podem torcer e autoamputar em até 10% dos pacientes, às vezes com hemorragia volumosa. Normalmente solitários (70%) sem potencial de malignização.
Está indicado a colonoscopia após a retirada de pólipo juvenil no reto pelo risco da existência de vários pólipos (30%). Polipose juvenil, Síndrome de Cronkwhite-Canada e Síndrome de Peutez-Jeghers. Uma vez que nessas entidades os pólipos (hamartomas) podem ocorrer além do intestino grosso, no estômago e intestino delgado e guardam associação com neoplasias malignas; a terapêutica agressiva (cirurgias para ressecção intestinal) e um seguimento próximo são justificados.
A queixa mais comum é o sangramento retal, seguida pelo prolapso através do ânus e dor abdominal. Quando único e por não representar uma lesão pré-maligna, assim que removido, não se faz necessário seguimento posterior.
Neoplásicos (Risco de Câncer)
Adenomas
Neoplasia benigna caracterizada pela:
1. Perda do controle de crescimento epitelial;
2. Mitoses generalizadas em todas as camadas das criptas da mucosa;
3. Importante alteração na renovação celular;
4. Menor diferenciação celular;
5. Maior produção de muco.
Microscopicamente (histologia) são classificados em:
![]()
• Adenoma tubular. No mínimo 80% de sua estrutura são tubulares.

• Adenoma tubulo-viloso. Padrão misto, cada padrão representa mais de 20% de sua composição.
 • Adenoma viloso. No mínimo 80% de sua estrutura são digitiformes.
• Adenoma viloso. No mínimo 80% de sua estrutura são digitiformes.
Cada tipo é subdividido em displasia de baixo grau, displasia de alto grau e carcinoma “in situ”, demonstrando a evolução para o câncer, sequência adenoma-carcinoma.
Pólipo Serrilhado
Abriga tanto tecido neoplásico benigno (tecido adenomatoso) como tecido não neoplásico (hiperplásicos). Natureza epitelial com arquitetura serrilhada.
Adenoma Serrilhado Séssil
![]() Geralmente maior que 0,5 cm, plano ou séssil, localizado predominantemente à direita, podendo atingir mais de 2 cm. A lesão é difícil de detectar pela colonoscopia por geralmente ser plano ou séssil, da mesma cor da mucosa que o rodeia e o muco que o cobre apagar o seu padrão vascular. O câncer colorretal originado do adenoma serrilhado plano ou séssil apresenta histologia mucinosa, e se diferencia da Síndrome de Lynch por ocorrer em pacientes com mais de 65 anos e por ser derivado do pólipo serrilhado ao invés do adenoma.
Geralmente maior que 0,5 cm, plano ou séssil, localizado predominantemente à direita, podendo atingir mais de 2 cm. A lesão é difícil de detectar pela colonoscopia por geralmente ser plano ou séssil, da mesma cor da mucosa que o rodeia e o muco que o cobre apagar o seu padrão vascular. O câncer colorretal originado do adenoma serrilhado plano ou séssil apresenta histologia mucinosa, e se diferencia da Síndrome de Lynch por ocorrer em pacientes com mais de 65 anos e por ser derivado do pólipo serrilhado ao invés do adenoma.
Adenoma Serrilhado Tradicional
Geralmente pediculado, exibe arquitetura complexa serrilhada, podendo ser confundido com ![]() adenoma viloso. Estes pólipos devem ser ressecados completamente sempre que possível. Apenas recentemente estudos avaliaram o risco dos pólipos serrilhados estratificando-os pelo tamanho, número, presença de displasia e morfologia. Não existem estudos de seguimento longo.
adenoma viloso. Estes pólipos devem ser ressecados completamente sempre que possível. Apenas recentemente estudos avaliaram o risco dos pólipos serrilhados estratificando-os pelo tamanho, número, presença de displasia e morfologia. Não existem estudos de seguimento longo.
Pólipo Serrilhado Misto
Pólipo misto com componente convencional (adenomatoso) e componente serrilhado (pólipo hiperplásico, adenoma serrilhado séssil ou adenoma serrilhado tradicional) intimamente misturados. Mais frequentes e em grande quantidade nas poliposes colônicas.
Sequência Adenoma-Carcinoma e Pólipo Serrilhado-Carcinoma
Evidências indiretas clínicas e epidemiológicas das sequências:
1. A prevalência do câncer colorretal segue a distribuição geográfica dos pólipos, isto é, o seguimento do cólon onde o pólipo é mais frequentemente encontrado é o mesmo do câncer;
2. O câncer colorretal surge cerca de cinco anos após o surgimento dos pólipos, isto é, em faixas etárias maiores;
3. Cerca de um terço dos intestinos retirados com câncer colorretal incluem um ou mais pólipos associados (a frequência é seis vezes maior que a encontrada nos intestinos retirados para outras doenças;
4. O tamanho do adenoma é o principal fator de risco para o surgimento da displasia de alto grau e o câncer.
Evidências indiretas de estudos clínicos das sequências:
 1. Os adenomas e serrilhados colorretais são encontrados em cerca de 30% das colonoscopias em pacientes acima dos 45 anos.
1. Os adenomas e serrilhados colorretais são encontrados em cerca de 30% das colonoscopias em pacientes acima dos 45 anos.
2. O tamanho do adenoma é o principal fator de risco para o encontro da displasia de alto grau, que invariavelmente evolui para o câncer colorretal.
3. Cerca de 30% dos adenomas vilosos possuem áreas de displasia de alto grau (30%), o que explica o maior risco de evoluir para o câncer colorretal.
4. O tempo médio de transformação para o câncer é de 7 anos para os pólipos com displasia de alto grau e de 11 anos para os adenomas de baixo grau.
5. Os estudos que analisaram a morfologia dos adenomas encontraram diferenças importantes na prevalência da displasia de alto grau e do câncer colorretal. Encontrada em 75% dos adenomas deprimidos, em comparação com 14,3% e 8,3% dos adenomas planos e polipoides, respectivamente.
Sequência Adenoma-Carcinoma 
Responsável por 80% dos cânceres colorretais esporádicos.
O primeiro evento é a inativação do gene APC o que leva a um crescimento celular anormal e por múltiplas vias de carcinogênese, progride para carcinoma invasivo.
Sequência Pólipo Serrilhado-Carcinoma
Responsável por 15% dos cânceres colorretais esporádicos.
Evidências indiretas de estudos clínicos:
1. Recentes estudos moleculares fornecem evidências convincentes da evolução do adenoma serrilhado séssil ou plano, mais frequente no cólon direito e do adenoma serrilhado tradicional geralmente pediculado ou subpediculado, mais frequente no cólon esquerdo para o carcinoma serrilhado do cólon direito e do cólon esquerdo respectivamente. Do adenoma serrilhado misto quando existe a sobreposição de ambos e presentes tanto no colon direito quanto no cólon esquerdo.
2. Pólipo serrilhado residual é encontrado em cerca de 5,8% dos cânceres colorretais.
3. O risco de câncer colorretal na polipose serrilhada varia de 20% a 50%.
Polipose serrilhada: mínimo de 5 pólipos serrilhados proximais ao sigmoide, com 2 ou mais ≥ a 10 mm; mais de 20 pólipos serrilhados de qualquer tamanho e qualquer pólipo serrilhado proximal ao sigmoide em pacientes com história familiar de polipose serrilhada.
Responsável por 5% dos cânc![]() eres colorretais esporádicos. (Lancet 2000, 355(9211):1211-1214).
eres colorretais esporádicos. (Lancet 2000, 355(9211):1211-1214).
1- Câncer colorretal sem componente adenomatoso, serrilhado ou “in situ”. Geralmente superficiais ou deprimidas que mesmo pequenos (≤ 10 mm) são avançados.
2- Raros e pouco se sabe sobre as características biológicas e a história natural. Encontrado com maior frquência no oriente, especialmente no japão.
3- Mais frequentes no cólon direito (85%), mas raramente são encontrados em pacientes pós colonoscopia (câncer de intervalo).
Síndromes hereditárias associadas ao câncer colorretal
Atualmente está bem estabelecido que existem doenças hereditárias relacionadas ao câncer colorretal. Dentre elas estão o Câncer Colorretal Hereditário Sem Polipose (HNPCC = “Hereditary Nonpoliposis Colorectal Cancer”) e Polipose Adenomatosa Familiar (FAP). Ambas são doenças hereditárias, ou seja, que são transmitidas de pais para filhos, caracterizadas pela presença de vários casos de câncer colorretal na família.
Estas duas síndromes apresentam um padrão de herança autossômico dominante, o que significa que os indivíduos afetados são geneticamente heterozigotos e que os descendentes de um paciente afetado têm 50% de chance de ser também portador da mutação, assim como cada progenitor tem 50% de possibilidade de transmitir o alelo mutado. Apenas as crianças homozigotas recessivas (25%) não desenvolverão a doença e não poderão transmiti-la. Entretanto, aproximadamente um terço das pessoas afetadas não possui nenhum parente com a mesma desordem. Esses indivíduos têm uma nova mutação no gene APC, podendo, obviamente transmiti-la para sua prole.
Polipose Adenomatosa Familiar ou FAP 
Corresponde a 1% dos casos de câncer colorretal. Centenas a milhares de pólipos adenomatosos desenvolvem-se entre os 7 e os 36 anos (média de idade de 16 anos). Aos 35 anos, 95% dos indivíduos com a síndrome possuem pólipos, sendo que, se não for feita colectomia total, o câncer colorretal é inevitável. A média de idade em que este evento ocorre, ou seja, o surgimento do câncer é de 39 anos (entre 34 e 43 anos, geralmente).
Manifestações extracolônicas
Podem também estar presentes, incluindo-se pólipos no estômago e duodeno, osteomas, anormalidades dentárias, hipertrofia congênita do epitélio pigmentar da retina, tumores de tecidos moles, tumores desmóides e outros cânceres associados, tais como tumores do SNC, da tireóide e hepatoblastomas.
Mutação
O gene APC no locus cromossômico 5p21 encontra-se mutado na FAP. O gene específico no cromossomo 5, sítio da mutação responsável, é chamado de gene APC.
Sintomas
A maioria desenvolvem os pólipos de forma assintomática. Entretanto, alguns sintomas podem ocorrer:
ᴥ» Presença de sangue rutilante acompanhando as evacuações;
ᴥ» Períodos de diarreia e/ou constipação não explicados pela dieta ou por viroses;
ᴥ» Dor em cólica na região gástrica;
ᴥ» Sensação frequente de distensão abdominal;
ᴥ» Perda de peso persistente e não explicada;
ᴥ» Astenia.
As características clínicas das lesões malignas, por sua vez, são aquelas comuns a todas as neoplasias semelhantes adquiridas, tais como perda de peso, inanição, obstrução intestinal ou diarréia hemorrágica.
Polipose Adenomatosa Familiar Atenuada
A FAP atenuada (FAPA) é caracterizada pelo desenvolvimento de menos de 100 pólipos (simultâneos ou não) no cólon e reto. A incidência é desconhecida, mas acredita-se que seja tão rara quanto a clássica.
A média de idade ao diagnóstico de câncer de cólon é de 50 a 55 anos, 10 a 15 anos mais tarde do que a doença clássica, porém mais cedo do que a população não afetada que desenvolve neoplasia de cólon esporadicamente.
Pólipos e câncer no trato gastrintestinal alto podem também ser vistos em pessoas com FAP atenuada e, embora as manifestações extra intestinais possam estar presentes, tumores desmoides e lesões por HCEPR (hipertrofia congênita do epitélio pigmentar da retina) são raros.
São frequentemente encontrados em famílias que também possuem indivíduos com FAP clássica, e a forma atenuada associa-se com uma mutação variante no gene APC em alguns casos.
A FAP atenuada, que apresenta distribuição mais proximal dos adenomas se comparada à forma clássica, sendo que alguns são sésseis e outros podem ser não-polipoides ou planos.
Testes genéticos para detecção do APC mutado podem ser um componente importante na avaliação dos pacientes em que se suspeita da doença, sendo que os métodos diagnósticos são os mesmos utilizados para a FAP clássica.
Síndrome de Gardner (SG)
Síndrome de Gardner é a associação de polipose adenomatosa colônica (e às vezes, polipose gástrica ou do intestino delgado) a osteomas craniofaciais e tumores de tecidos moles (cistos epidermóides, fibromas e tumores desmóides), além de HCPER- Hipertrofia congênita do epitélio pigmentar da retina (é possível, inclusive, que essas lesões sejam as únicas manifestações da síndrome).
Síndrome de Turcot (ST)
A síndrome de Turcot é rara e caracteriza-se clinicamente pela ocorrência de um tumor cerebral primário e de múltiplos adenomas colorretais. Os tumores do Sistema Nervoso Central costumam ser malignos (SNC), usualmente meduloblastomas ou, com uma incidência um pouco inferior, glioblastomas multiformes. Quanto aos pólipos, estes se encontram presentes em menor quantidade do que na FAP (entre 20 e 300, normalmente).
Diagnóstico da Polipose Adenomatosa Familiar
Em pacientes com sintomas, a retossigmoidoscopia rígida ou flexível permite o diagnóstico dos pólipos e a retirada de alguns para exame. Em pacientes com história familiar de FAP, na indisponibilidade do teste genético, realiza-se a retossigmoidoscopia rígida ou flexível no início da adolescência e caso negativo, repeti-los anualmente permite o diagnóstico. Nestes casos o não encontro de pólipos entre os 18 e 20 anos de idade, a colonoscopia anual está indicada para a pesquisa da forma atenuada, quando os pólipos podem ser proximais e não alcançáveis pelos primeiros exames. Em pacientes sem sintomas apenas o acaso permite o diagnóstico precoce da doença.
Teste Genético
O teste de genética molecular APC detecta até 95% das mutações causadoras da doença. Está indicado no diagnóstico precoce de membros com 11 anos ou mais de famílias de alto risco. Na confirmação de diagnóstico de FAP em pacientes com achados equivocados. Indivíduos com menos de 100 pólipos adenomatosos e um diagnóstico duvidoso de FAP. Quando é identificada uma mutação em uma família, a pesquisa da mesma em outros membros pode ser efetuada rápida, precisa e economicamente. Os testes são tipicamente realizados no DNA extraído de leucócitos de uma amostra de sangue. Os principais são o sequenciamento do gene e o teste da proteína truncada (PTT).
Consequências do teste genético positivo
1- No caso de uma família que claramente possui história de FAP, haverá indivíduos afetados e indivíduos em risco.
2- Assim, os que não possuírem a mutação não mais precisarão participar do intensivo programa de rastreamento, enquanto os portadores de mutação devem seguir com o rastreamento e serem indicados para a cirurgia profilática.
Consequências do teste genético negativo
1- Falha em reduzir a incerteza, pois os testes não possuem 100% de sensibilidade e especificidade, podendo gerar resultados ambíguos.
2- Dano psicológico causado pela notícia de resultado positivo no teste de DNA, podendo ser acompanhado por sentimentos de raiva, ansiedade e depressão.
3- Discriminação genética, principalmente na área social, casamento, saúde e emprego.
4- Confiança entre o paciente e o médico, devido ao medo da discriminação. 5- Questões éticas, entre outras.
Tratamento da Polipose Adenomatosa Familiar
O tratamento, enfim, é normalmente feito pela colectomia total com ileorretoanastomose ou proctocolectomia total com ileoanastomose definitiva. No primeiro caso, é necessária a fulguração dos pólipos localizados no reto, uma vez que não haja evidência de transformação maligna. O paciente deve ser revisto com intervalos de 6 meses para afastar a possibilidade de formação de novos pólipos com potencial maligno. Atualmente, em casos selecionados, vem sendo adotada a retirada total do intestino grosso com anastomose de uma bolsa ileal (reservatório ileal) ao canal anal.
Risco para os membros da família
Pais de um portador da FAP Aproximadamente 75 a 80% dos indivíduos com FAP têm um dos pais afetado. Portanto, sempre é apropriado avaliar seus pais com teste molecular do gene APC. Aproximadamente 20 a 25% de indivíduos com FAP têm o gene alterado como resultado de uma mutação genética nova.
Irmãos de um portador da FAP O risco para os irmãos depende da condição genética dos pais. Se um dos pais é afetado, o risco é de 50%. Se nenhum dos pais de um indivíduo com FAP se encontra nos critérios clínicos para a doença, o risco de os irmãos de um indivíduo afetado terem FAP é o risco populacional para essa desordem.
Descendentes de um portador da FAP Todos os filhos de um indivíduo com FAP têm uma chance de 50% de herança da mutação.
Outros membros da família de um portador da FAP O risco para os outros membros da família depende da condição dos pais do probando. Se um dos pais encontra-se afetado, os integrantes de sua família estão sob risco.
Câncer Colorretal Hereditário Não Polipose (HNPCC)
Também conhecido como Síndrome de Lynch, a HNPCC é uma doença autossômica dominante responsável por cerca de 5% a 10% do total dos cânceres colorretais. É a síndrome hereditária mais comum de predisposição ao câncer colorretal. Ainda que os critérios para diagnóstico de Amsterdam e tratamento estejam estabelecidos, o conhecimento da síndrome pela classe médica é insuficiente.
Se comporta como na FAP, isto é, têm 50% de chance de passar a condição para o seu filho. Independe da atuação de fatores ambientais. Os indivíduos com HNPCC têm uma probabilidade próxima dos 70% de desenvolver o câncer colorretal. Ocorre geralmente em idades mais jovens, os tumores localizam-se principalmente no cólon direito. Apresentam ainda, um risco aumentado de tumores extra-cólicos (TEC), principalmente carcinomas do endométrio, ovário, estômago, intestino delgado, urotélio, pâncreas e vias biliares.
Critérios de Amsterdam
Pelo menos três casos de câncer colorretal (CCR), que preencham os seguintes critérios:
1- Um membro seja parente em 1º grau dos outros dois.
2- Pelos menos 2 gerações sucessivas acometidas.
3- Pelo menos um dos casos de câncer colorretal diagnosticado abaixo dos 50 anos.
4- Polipose adenomatosa familiar deve ser excluída.
Os critérios de Amsterdam (CA), embora altamente específicos, têm valor muito limitado, inclusive para nossa população, pois são extremamente restritivos. Assim, foram propostos os critérios de Bethesda, menos restritivos, com o intuito de levantar a suspeita de HNPCC e a utilização de testes laboratoriais, como a instabilidade de microssatélites, para auxiliar a definir um maior número de famílias de risco para HNPCC.
Critérios de Bethesda
1- Indivíduos com câncer que preencham os critérios de Amsterdam.
2- Indivíduos com 2 cânceres relacionados com HNPCC, incluindo câncer colorretal metacrônicos e sincrônicos ou cânceres extracólicos associados.
3- Indivíduos com câncer colorretal e um parente de 1º grau com câncer colorretal ou câncer extracólico relacionado com HNPCC e/ou adenoma colorretal (um dos cânceres diagnosticado abaixo dos 45 anos e os adenomas abaixo dos 40 anos).
4- Indivíduos com câncer colorretal ou endometrial diagnosticado abaixo dos 45 anos.
5- Indivíduos com câncer de cólon direito com padrão histológico indiferenciado abaixo dos 45 anos.
6- Indivíduos com câncer colorretal com células em anel de sinete diagnosticado abaixo dos 45 anos.
7- Indivíduos com adenomas diagnosticados antes dos 40 anos.
8- Cânceres extracólicos associados: endométrio, ovário, estômago, hepatobiliar, intestino delgado, células de transição da pelve renal ou ureter.
Teste Genético para HNPCC
O diagnóstico de HNPCC pode ser realizado de duas maneiras: clinicamente, através do preenchimento dos critérios de Amsterdam; e geneticamente, pela identificação de mutações decorrente da instabilidade de microssatélites (MSI), fruto da inativação das proteínas de reparo do DNA. Mutações germinativas nos genes hMLH1 e hMSH2, dois dos principais genes do complexo MMR, estão presentes em 80% a 90% dos casos de HNPCC. 
Embora isso pareça simples, na prática os médicos encontram enormes dificuldades para estabelecer tal diagnóstico, seja porque não dispõem de informações familiares adequadas com relação ao histórico do paciente, seja porque o sequenciamento genético não é ainda uma realidade na prática médica.
Protocolo de rastreamento de famílias em risco para HNPCC
Seleciona as famílias através dos critérios de Bethesda, seguido da investigação genética com a execução do teste de instabilidade de microssatélites (MSI) e/ou a imunohistoquímica para proteínas dos genes de reparo do DNA em amostras do câncer ressecado por cirurgia ou colonoscopia. Uma vez que um destes testes mostra-se alterado, passa para a pesquisa de mutações por sequenciamento nos genes de reparo do DNA.
A diferenciação dos casos de HNPCC daqueles de câncer colorretal esporádico visa o aconselhamento genético, programas específicos de rastreamento e encaminhamento da família para análise mutacional no complexo MMR.
Síndrome de Peutz-Jeghers (SPJ)
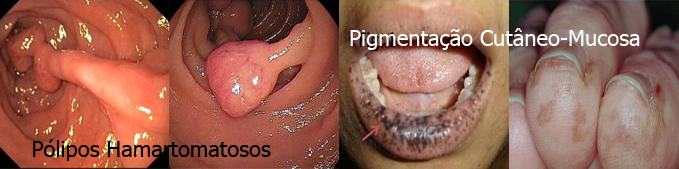 É uma síndrome herdada de forma autossômica dominante e se caracteriza clinicamente pela associação de pigmentação melanótica cutâneo-mucosa com pólipos hamartomatosos encontrados no trato digestivo (principalmente no intestino delgado) e, ocasionalmente, no trato urinário e respiratório. As manchas aparecem frequentemente na cavidade oral, regiões perioral e periorbitária, dedos das mãos e pés ou genitália, podendo variar em coloração.
É uma síndrome herdada de forma autossômica dominante e se caracteriza clinicamente pela associação de pigmentação melanótica cutâneo-mucosa com pólipos hamartomatosos encontrados no trato digestivo (principalmente no intestino delgado) e, ocasionalmente, no trato urinário e respiratório. As manchas aparecem frequentemente na cavidade oral, regiões perioral e periorbitária, dedos das mãos e pés ou genitália, podendo variar em coloração.
Os sintomas da SPJ aparecem logo na primeira década de vida. Manifestações variadas com episódios repetidos de dor abdominal, sangramento intestinal inexplicado, prolapso de pólipo retal, irregularidades menstruais e puberdade precoce (devido ao hiperestrogenismo de tumores de cordões sexuais), ginecomastia, crescimento acelerado (tumores de células de Sertoli) e massa testicular podem estar presentes.
As principais causas de morbi-mortalidade ocorrem tipicamente na segunda década de vida e são representadas por intussuscepção de intestino delgado (43%), dor abdominal (23%), hematoquezia (14%), prolapso de pólipo colônico (7%) e presença de neoplasia. Noventa por cento dos pacientes apresentam anemia associada a dor abdominal em cólica, recorrente, a qual é decorrente de intussuscepções transitórias (e muitas vezes, reversíveis).
Nessa síndrome há um risco aumentado de ocorrência de carcinoma em diversos órgãos, sendo mais acometidos o pâncreas (30%), a mama (25%), ovário e útero (20%), testículo (10%), estômago e intestino delgado (10%)2. Aproximadamente 50% dos pacientes desenvolverão câncer até os 57 anos3. Dessa forma, é recomendado o seguimento destes pacientes com exames de imagem e endoscópicos. Alguns protocolos recomendam endoscopia digestiva alta e retossigmoidoscopia a cada dois a cinco anos até os 20 anos de idade. Em idades mais avançadas a colonoscopia deve ser acrescentada.
O tratamento cirúrgico está indicado principalmente nos casos de obstrução, sangramento e intussuscepção. A tendência é que estes procedimentos sejam conservadores (polipectomias ou ressecções segmentares). Como os pólipos se distribuem por todo o trato digestivo, qualquer tentativa de ressecções mais alargadas e extensas não terá êxito no controle da doença, além de piorar o estado nutricional destes pacientes.
O método endoscópico, além de permitir o diagnóstico e localização das lesões, tem um papel importante na terapêutica possibilitando a ressecção de pólipos isoladamente. Assim, é possível estudar e manipular o trato gastrointestinal, restringindo enterectomia aos segmentos com complicações.
Polipose Juvenil (PJ) 
A Polipose Juvenil é uma síndrome autossômica dominante. É uma condição incomum e afeta de 1 em 100.000 a 1 em 160.000 pessoas e costuma se manifestar entre 4 e 14 anos de idade. O seu diagnóstico é feito quando os seguintes critérios clínicos são encontrados:
1- Mais de 5 pólipos juvenis no cólon ou no reto, ou;
2- Pólipos juvenis em outras áreas do trato gastrointestinal, ou;
3- Qualquer número de pólipos juvenis e uma história familiar positiva.
A Polipose Juvenil é classificada em três subgrupos:
1- Polipose Juvenil da infância é associada a sangramento gastrointestinal de repetição, prolapso retal e intussuscepção, que ocasionam a morte precoce dos pacientes;
2- Polipose Juvenil colônica é a forma mais comum; os pólipos localizam-se no cólon e os portadores têm um prognóstico bom;
3- Polipose gastrointestinal juvenil generalizada quando a distribuição dos pólipos se assemelha à verificada na Síndrome de Peutz-Jeghers, mas a pigmentação da mucosa oral, lábios e dedos está ausente;
Os pólipos na Polipose Juvenil são hamartomatosos e variam de poucos milímetros a 5 cm de diâmetro. São revestidos por uma única camada de epitélio de células colunares, que frequentemente ulceram, levando à hemorragia e à infecção.
A maioria dos pólipos grandes são pediculados e responsável pela evolução natural e pelos sintomas recidivantes da polipose juvenil, que incluem hemorragia gastrointestinal, dor abdominal, obstrução intestinal, anemia, intussuscepção, prolapso, enteropatia perdedora de proteínas, desnutrição e auto-amputação dos pólipos. Manifestações extra colônicas incluem: macrocefalia, alopecia e fenda palatina. Nenhum dos pacientes apresentou manifestações extra colônicas.
Vários trabalhos na literatura relatam a ocorrência de câncer gastrointestinal em pacientes portadores de Polipose Juvenil.
Os pacientes portadores da Polipose Juvenil devem ser acompanhados rigorosamente devido ao alto índice de recidiva dos pólipos. Exames endoscópicos e excisão dos pólipos encontrados devem ser realizados regularmente para regressão das manifestações clínicas e prevenção de transformações neoplásicas. Quando os pólipos são numerosos e difíceis de controlar endoscopicamente, quando sintomas como, sangramento retal e diarreia são abundantes, ou quando há alguma suspeita de câncer colorretal, os pacientes devem ser submetidos à cirurgia.
O tratamento cirúrgico indicado envolve colectomia total ou proctocolectomia total restaurativa com bolsa ileal. Mesmo após o procedimento cirúrgico, é indicado acompanhamento endoscópico em virtude da possibilidade de surgimento de outros pólipos no trato gastrointestinal restante.
O tratamento medicamentoso da Polipose Juvenil ainda permanece controverso.
Síndrome Polipóide Hereditária Mista (SPHM)
Apresenta um padrão de herança genética desconhecido. A síndrome é caracterizada por pólipos atípicos, que contêm histologia mista, ou por múltiplos pólipos de mais de um tipo histológico em um mesmo indivíduo. É uma variante da polipose juvenil caracterizada por pólipos adenomatosos e hamartomatosos.
Neurofibromatose tipo 1 (NF1)
Embora mundialmente conhecida como “doença de von Recklinghausen”. A NF-1 afeta um em cada 2.000 a 4.000 nascidos vivos, sendo a mais freqüente das neuromesoectodermoses, doenças genéticas de herança geralmente autossômica dominante, nas quais se observa associação de alterações do sistema nervoso central e da coloração da pele. A NF-1 tem herança autossômica dominante com penetrância completa e praticamente metade dos casos deve-se a mutação nova. Por se tratar de uma doença que compromete os três folhetos germinativos sua apresentação clínica é variada e pode, teoricamente, afetar todos os órgãos e sistemas.
A neurofibromatose é classificada em tipo 1 (forma periférica) e tipo 2 (forma central). Um terceiro tipo frequentemente citado é a forma segmentar, na qual a doença é restrita à determinada região do corpo. Com base em dados clínicos, radiológicos e histológicos, foram estabelecidos critérios para o diagnóstico da NF-1 pelo NIHCDC, sendo necessária a presença de pelo menos 2 dos seguintes critérios:
1- Seis ou mais manchas café com leite com diâmetro superior à 5 mm em pacientes menores de 6 anos;
2- Seis ou mais manchas café com leite com diâmetro superior à 15 mm em pacientes maiores de 6 anos;
3- Sardas inguinais e/ou axilares;
4- Glioma de nervo óptico;
5- Dois ou mais neurofibromas de qualquer tipo ou um neurofibroma plexiforme;
6- Dois ou mais hamartomas de íris (nódulos de Lisch);
7- Lesões ósseas distintas como displasia do osso esfenóide e/ou afilamento da córtex de ossos longos com ou sem pseudo-artrose;
7- Parente de primeiro grau com NF1 diagnosticada de acordo com os critérios anteriormente citados.
Além das anteriormente citadas, diversas outras manifestações clínicas são descritas em pacientes com NF-1, destacando-se as neurológicas (retardo mental, comportamento autista, dislexia, transtorno do déficit de atenção/hiperatividade, polineuropatia, distúrbios do sono e malformações cerebrais), endocrinológicas (hiperparatireoidismo, hiperprolactinemia, irregularidades menstruais, menopausa precoce), cardiovasculares (hipertensão arterial sistêmica e malformações cardíacas em graus variados de severidade), do sistema gastrintestinal e do trato geniturinário.
Síndrome de Cronkite-Canadá
É uma polipose hamartomatosa gastrointestinal generalizada associada à hiperpigmentação cutânea, perda de cabelo e atrofi a ungueal. Associa-se a diarreia crônica e desnutrição com hipoproteinemia.
a ungueal. Associa-se a diarreia crônica e desnutrição com hipoproteinemia.
Apresenta múltiplos pólipos (hamartoma) no intestino delgado e cólon,também no estômago, mas não em esôfago. Os pólipos são sésseis (não pediculados) com erosões superficiais. É uma doença muito frequente, pelo menos nos países ocidentais, levando a maioria dos casos publicados de países asiáticos.
A causa é desconhecida. Dados sugere uma doença com uma base inflamatória que pode ser influenciado de alguma forma por agentes bacterianos. Recentemente, foi publicada uma possível relação à infecção por Helicobacter pylori.
Tratamento – Tem sido descrito remissões da doença após melhorar o estado nutricional e restaurar os distúrbios electrólitos desses pacientes, bem como a utilização da associação de drogas como os antibióticos (ampicilina, tetraciclina, metronidazol, ciprofloxacina , etc), corticóide oral e intravenosa, antagonistas H1 e H2 do receptor da histamina (loratadina e ranitidina), medicamentos antidiarreicos ou cromoglicato dissódico. Não há dúvida sobre a necessidade de tratamento nutricional, oral ou parentérica, bem como o controlo de iônico e traço deficiência elementos. Prognóstico – É uma doença com mau prognóstico. A maioria dos pacientes morrem por causa de um quadro de desnutrição grave, frequentemente complicado com infecções.
Hiperplasia nodular linfóide
É uma desordem linfoproliferativa que resulta em nódulos linfóides hiperplásicos no intestino delgado, estômago e cólon, podendo estar relacionada com síndromes de imunodeficiência variadas.
Carac![]() teriza-se por nódulos submucosos, que representam uma hiperplasia do tecido linfóide, podendo aparecer em diversos segmentos do intestino.São pólipos pequenos, de 1-5 mm, sésseis, umbilicados, predominando na porção distal do cólon.
teriza-se por nódulos submucosos, que representam uma hiperplasia do tecido linfóide, podendo aparecer em diversos segmentos do intestino.São pólipos pequenos, de 1-5 mm, sésseis, umbilicados, predominando na porção distal do cólon.
Os aspectos radiológicos e endoscópicos podem levar a confusão com síndromes de FAP. A diferenciação se dá pela biópsia, que mostra que esses pequenos pólipos são compostos por nódulos linfóides aumentados, com centros germinais proeminentes na lâmina própria e na superfície da submucosa.
Pode haver dor abdominal, náusea, hiporexia, emagrecimento, má absorção, sangue oculto fecal e anemia.
Em pacientes com imunodeficiência associada, os nódulos estão limitados ao intestino delgado, enquanto que nos imunocompetentes, estes localizam-se também no cólon.
A aparição destes nódulos pode representar uma resposta do tecido linfóide intestinal a estímulos infecciosos, químicos e traumáticos (giardiase, reações adversas aos alimentos, etc).
Alguns trabalhos sugerem que poderia existir um risco aumentado de linfoma no intestino delgado nestes pacientes, porém, o risco ainda é desconhecido, sendo portanto uma entidade benigna e autolimitada.
Polipose inflamatória
É uma moléstia adquirida onde pólipos não neoplásicos. Trata-se de pólipos irregulares, solitários, localizados no cólon e intestino delgado, geralmente com menos de 10 mm.
Também chamados de pseudopólipos, que podem ser observados em todas as variedades de colite aguda, durante a fase de regeneração e cicatrização.
Podem ser observados na Doença de Crohn, na colite isquêmica, na amebíase, esquistossomose e nas infecções bacterianas crônicas.
O tratamento é o da doença de base.
Síndrome de Cowden
A síndrome de Cowden(SC) ou síndrome de múltiplos hamartomas(SMH) é rara e de herança autossômica dominante e expressividade variável.
É caracterizada por múltiplas lesões hamartomatosas de origem ectodérmica, mesodérmica e endodérmica. O órgão mais acometido é a pele, e as lesões mucocutâneas estão presentes em proporção que varia de 99 a 100% dos casos, e incluem hiperceratose dos lábios, língua e narinas. Aspecto semelhante ao de pequenas “pedras arredondadas”.
Os pólipos podem se localizar em qualquer segmento do TGI, como cólon, retossigmóide, estômago, duodeno, intestino delgado e esôfago, em ordem decrescente de acometimento e o ânus também pode ser afetado. São tipicamente pequenos, múltiplos e sésseis. O quadro histológico das lesões gastrointestinais pode ser hamartomatoso, lipomatoso, linfomatoso, inflamatório, hiperplásicoe, ocasionalmente, adenomatoso.
Esses sinais precedem o desenvolvimento do câncer em vários anos, servindo como importantes marcadores clínicos na identificação de pacientes com alto risco para desenvolver câncer de mama (lesões fibrocísticas ou fibroadenomatosas) e tireoide.
O sistema nervoso pode ser acometido de diversas maneiras: ganglioneuromase neurofibromas sintomáticos ou assintomáticos, que podem ser encontrados incidentalmente ou em associações com lesões cutâneas.
Um terço dos pacientes tem defeitos esqueléticos, entre eles, aumento da circunferência do crânio que caracteriza um dos achados extracutâneosmais comuns. Outras alterações esqueléticas encontradas são fácies adenoide, cifose, cifoescoliose, pectusexcavatum, mãos e pés grandes e sindactilia.
Em relação ao sistema cardiovascular, verificam-se hipertensão, defeitos no septo atrial, prolapsoda válvula mitral e insuficiência aórtica e mitral.
Na avaliação do trato gastrointestinal recomenda-se a endoscopia digestiva alta, que pode demonstrar polipose esofagiana, gástrica e duodenal, além de hérnia de hiato e esofagite erosiva grave.
A colonoscopia pode visualizar pólipos em todo o cólon e reto. Devido a associações com malignidades internas o diagnóstico precoce é essencial.
O tratamento da é controvertido e visa principalmente à melhora do aspecto estético das lesões e à busca ativa de neoplasias associadas.
A excisão de pólipos, com fulguração da base, não erradica essas lesões e ocorre a recorrência de lesões benignas nos mesmos sítios dos neoplasmas anteriores.
Todos os pacientes devem ser rigorosamente submetidos à busca de malignidades ocultas, com ênfase para mama, glândula tireoide, trato gastrointestinal e sistema nervoso.
O reconhecimento precoce e o manuseio dessas associações podem reduzir a morbidade e mortalidade presentes nesta síndrome. Até o momento, não há recomendações gerais para o seguimento destes pacientes.
Pólipo com Câncer Colorretal (maligno)
Câncer intramucoso ou “in situ”
Não invade o plano da muscular da mucosa e não apresentam riscos de metástases.
A ressecção endoscópica completa cura o paciente e, portanto não é necessário cirurgia em nenhum dos casos.
Câncer invasivo
Invade além da lâmina própria e ultrapassam o plano da muscular da mucosa.
A retirada endoscópica pode ser curativa e depende da análise de cada caso. O receio se deve ao risco da metástase quando a lesão atinge a submucosa.
Geralmente o pólipo maligno é retirado pela colonoscopia e o exame histológico (anatomopatológico) evidencia o foco de câncer e define os riscos na qual a conduta médica é determinada.
Classificação proposta por Haggitt (Haggitt, 1985) para o câncer colorretal precoce. Um pólipo é considerado pediculado quando o comprimento da haste é maior do que o seu diâmetro (Wilcox e Beck, 1987).
Nessa classificação, definem-se 4 níveis de invasão se o pólipo for pediculado e todos os pólipos sésseis que abrigam carcinoma invasivo são classificados como nível 4 de invasão.
ᴥ» Nível 0 cor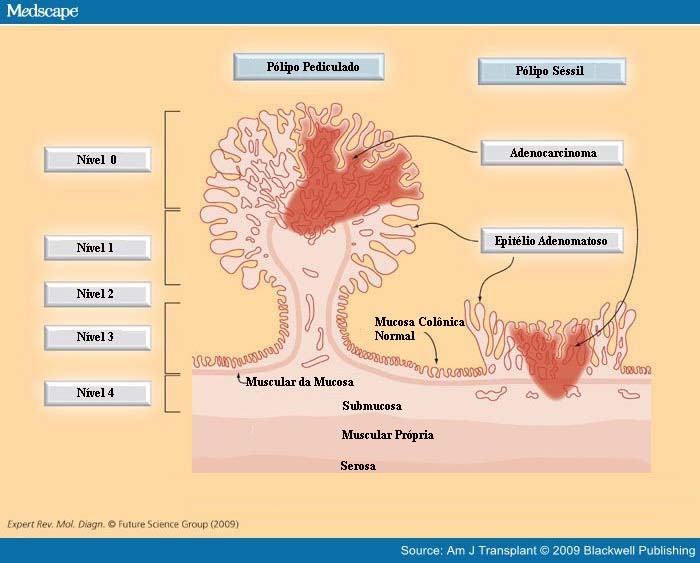 responde ao carcinoma in situ;
responde ao carcinoma in situ;
ᴥ» Nível 1 corresponde a invasão da muscular da mucosa limitada à cabeça do pólipo;
ᴥ» Nível 2 corresponde à invasão do colo do pólipo (o colo é definido como a região do pedículo onde se dá a transição entre o componente adenomatoso e o revestimento epitelial normal);
ᴥ» Nível 3 corresponde à invasão da haste ou pedículo; e
ᴥ» Nível 4 corresponde à invasão da submucosa da parede intestinal na base do pólipo.
As lesões sésseis com câncer invasivo restrito à submucosa (T1) e nível 4 de Haggitt são subdividido em:
ᴥ» sm1 – invasão do terço mais superficial da submucosa, atingindo somente a camada muscular da mucosa;
ᴥ» sm2 – invasão do terço médio;
ᴥ» sm3 – invasão superficial da muscular própria.
A principal dificuldade dessa classificação é a significativa variabilidade intra e inter-observador associada à determinação do nível de invasão dentro de uma camada fina como a submucosa intestinal.
As lesões pediculadas, os níveis de invasão Haggitt 1, 2 e 3 correspondem a sm1 e o nível 4 se subdivide em sm1, sm2 ou sm3 conforme descrito acima.
Após a ressecção endoscópica com o anatomopatológico descrevendo o pólipo como maligno, cria uma pergunta a responder. O paciente está curado?
ᴥ» Câncer intramucoso (“in situ”) é considerado curado desde que a polipectomia ou mucosectomia seja adequada;
ᴥ» Câncer invasivo em qualquer situação não é considerado curado e a cirurgia clássica (colectomia segmentar) está indicada na maioria porque o risco de comprometimento linfonodal chega a 9%.
O risco de comprometimento linfonodal aumenta de acordo com a presença dos seguintes parâmetros anatomopatológicos:
ᴥ» Carcinoma indiferenciado ou pouco diferenciado;
ᴥ» Embolização linfática;
ᴥ» Embolização venosa;
ᴥ» Nível 4 de Haggitt;
ᴥ» Nível de invasão Sm3;
ᴥ» Margem de ressecção: menor do que 2 mm.
Os pólipos malignos são classificados de acordo com seu prognóstico em dois tipos:
Bom prognóstico: células bem diferenciadas, margens livres de tumor > 2 mm e ausência de invasão intravascular e linfática.
Nesses casos a recidiva tumoral gira em torno de 1,5%. Nos pacientes com alto risco cirúrgico (estado geral, idade avançada, e/ou doenças associadas) o procedimento endoscópico poderá ser considerada suficiente. Neste caso é importante tatuar com nanquim as proximidades e não o local da lesão, para não dificultar a identificação de possível recidiva. Colonoscopia de seguimento varia de 1 a 3 anos.
Mau prognóstico: células indiferenciadas ou pouco diferenciadas, margem de ressecção menor que 2 mm e invasão intravascular e linfática.
Nesses casos o risco de tumor residual é alto, em torno de 9%. A colectomia segmentar oncológica está indicada. Neste caso é importante tatuar com nanquim as proximidades para que o cirurgião laparoscópico identifique com precisão o segmento a ser ressecado.
Pólipo serrilhado
ᴥ»Pólipos hiperplásicos
Vigilância – Em pacientes com pólipos hiperplásicos pequenos (<10 mm) confinados ao reto ou sigmóide, a colonoscopia de vigilância ou seguimento é recomendada em 5 a 10 anos, igual aos pacientes de risco médio.
ᴥ»Pólipos serrilhados sésseis e adenomas serrilhados tradicionais
Vigilância – O intervalo de vigilância é baseado no tamanho dos pólipos e na histologia.
1- Indivíduos com adenoma serrilhado séssil ou pediculado <10 mm de tamanho sem displasia são tratados como adenomas de baixo risco com uma primeira colonoscopia de vigilância em cinco anos.
2- Indivíduos com adenoma serrilhado séssil ou pediculado ≥ 10 mm, com displasia ou adenoma serrilhado tradicional são tratados como adenomas de alto risco com uma primeira colonoscopia de vigilância em três anos.
3- Outras recomendações de consenso sugeriram um acompanhamento colonoscópico mais precoce (intervalo de um a três anos) em indivíduos com dois ou mais adenomas serrilhado séssil ou pediculado ≥ 10 mm e naqueles com qualquer displasia.
●Síndrome da polipose serrilhada
Indicações para colectomia incluem:
ᴥ»Câncer colorretal documentado ou suspeito.
ᴥ»Pólipos com displasia de alto grau ou múltiplos adenomas maiores que 6 mm.
ᴥ»Aumentos no número de pólipos em exames consecutivos.
ᴥ»Incapacidade de examinar adequadamente o cólon por causa de múltiplos pólipos diminutivos.
ᴥ»Preferência do paciente para evitar a vigilância por colonoscopia.
Na ausência de evidências para orientar as recomendações do rastreamento para o câncer colorretal para os membros da família, sugiro o rastreamento de parentes de primeiro grau de indivíduos com síndrome da polipose serrilhada por volta dos 40 anos (ou 10 anos antes da idade mais precoce na apresentação da síndrome da polipose serrilhada na família). A vigilância subsequente é a cada cinco anos se nenhum pólipo for achado.
Pólipo adenomatoso (Adenoma)
Os pólipos colorretais adenomatosos são encontrados em cerca de 25% das pessoas até a idade de 50 anos; a prevalência continua a aumentar com o aumento da idade.
Avaliação do risco para câncer colorretal subsequente.
A avaliação do risco para a pessoa desenvolver câncer colorretal é importante para orientar a vigilância. Histologia, número e tamanho do adenoma são fatores de risco para adenoma e câncer metacrônico (por exemplo, diagnosticado seis meses ou mais após a remoção de um câncer ou adenoma). Em um acompanhamento médio de aproximadamente 13 anos, as taxas de ocorrência de câncer colorretal em indivíduos com adenoma avançado, adenoma não avançado e ausência de adenoma na colonoscopia basal foram de 20, 9 e 7 por 10.000 pessoas ao ano, respectivamente.
●Histologia – Pólipos adenomatosos com mais de 25% de componente viloso, displasia de alto grau ou câncer invasivo são fatores de risco para o desenvolvimento de câncer colorretal no futuro. Em alguns estudos, a vilosidade também prediz adenomas avançados no futuro.
Adenomas de baixo risco
Adenomas de baixo risco são definidos como um a dois adenomas tubulares <10 mm. A primeira colonoscopia de vigilância deve ser realizada em 3 a 10 anos. O momento da colonoscopia de vigilância subsequente é baseado nos achados da primeira colonoscopia de vigilância.
●Se não forem encontrados adenomas na primeira colonoscopia de vigilância, a próxima colonoscopia de vigilância é recomendada em 5 a 10 anos, mas somente se não existir outros fatores associados ao aumento do risco do câncer colorretal (por exemplo, câncer colorretal ou adenomas de alto risco em um parente de primeiro grau antes dos 60 anos de idade ou em dois parentes de primeiro grau, independentemente da idade)
●Se um adenoma de baixo risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos.
●Se um adenoma de alto risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 2 a 3 anos. Pacientes com um adenoma de alto risco em qualquer exame parece permanecer em alto risco e devem ter intervalos curtos de acompanhamento (por exemplo, 2 a cinco anos) para todas as colonoscopias de vigilância subsequentes.
Adenomas de alto risco
••Três ou mais adenomas.
••Adenoma avançado: adenoma tubular ≥ 10 mm; adenoma viloso e displasia de alto grau. Pacientes com um adenoma de alto risco em qualquer exame devem ter intervalos curtos de acompanhamento para vigilância.
●Primeira vigilância: pessoas com adenoma avançado (≥10 mm, histologia vilosa ou displasia de alto grau) ou entre 3 e 10 adenomas em sua colonoscopia devem ser submetidos a uma primeira colonoscopia de vigilância em 2 a 3 anos. Pacientes com mais de 10 adenomas devem ser avaliados para a síndrome de câncer colorretal hereditário e realizar colonoscopia de vigilância em 2 anos ou menos (veja as indicações para avaliação genética).
●Vigilância subsequente: o momento da colonoscopia de vigilância subsequente é baseado nos achados da primeira colonoscopia de vigilância.
¶¶Se não forem encontrados adenomas na primeira colonoscopia de vigilância, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos. Pacientes com um adenoma de alto risco em qualquer exame permanece como alto risco e devem ter intervalos de 2 a 3 anos de acompanhamento para todas as colonoscopias de vigilância subsequentes.
¶¶Se um adenoma de baixo risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos.
As recomendações assumem que a colonoscopia de base foi completa e adequada e que todos os pólipos visíveis foram completamente removidos.
O intervalo de 5 a 10 anos é apenas para os pacientes de risco básico, na ausência de outros fatores de risco associados ao aumento do risco de câncer colorretal (por exemplo, câncer colorretal ou adenoma de alto risco em um parente de primeiro grau antes dos 60 anos de idade ou dois parentes de primeiro grau em qualquer idade). * Se o resultado da segunda vigilância for negativo, não há evidências suficientes para fazer uma recomendação.
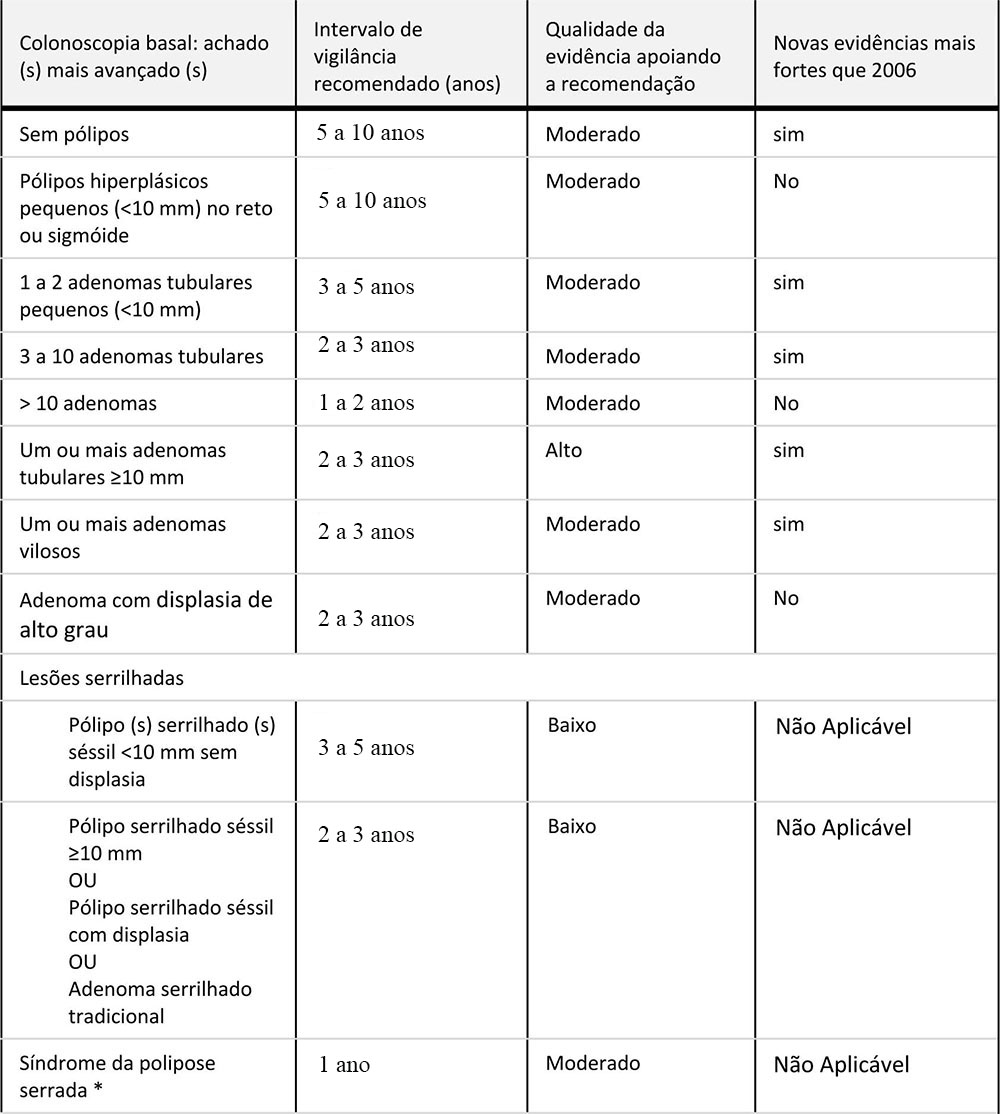 Recomendações para intervalos de vigilância e triagem do Câncer Colorretal em indivíduos com risco médio inicial.
Recomendações para intervalos de vigilância e triagem do Câncer Colorretal em indivíduos com risco médio inicial.![]() Recomendações para vigilância do câncer colorretal após a primeira colonoscopia de vigilância.
Recomendações para vigilância do câncer colorretal após a primeira colonoscopia de vigilância.
Indicações para avaliação genética
Aproximadamente 5 a 10 por cento dos cânceres colorretais são atribuíveis a uma síndrome hereditária de predisposição ao câncer. Uma síndrome hereditária de predisposição ao câncer colorretal deve ser considerada em pacientes que se apresentam com idade precoce no diagnóstico do adenoma ou câncer, ou com números ou histologias incomuns de câncer ou condições pré-malignas. Por exemplo, em pacientes com 10 ou mais adenomas colorretais cumulativos ou qualquer número de adenomas em combinação com adenomas duodenais ou ampulares, tumores desmóides, câncer de tireoide papilar, cistos epidérmicos e osteomas, deve levantar a possibilidade de polipose adenomatosa familiar (PAF).
Quando parar com a colonoscopia de prevenção?
Quando interromper a colonoscopia de prevenção do câncer colorretal em idosos? A interrupção deve ser considerada quando a expectativa de vida estimada for ≤ 10 anos.
Embora a colonoscopia de prevenção reduza em 50% a incidência do câncer colorretal em idosos (≥ 75 anos), em determinado momento ela deixa de aumentar significativamente a expectativa de vida e, portanto, não deve ser oferecida. Existem evidências e consensos claros sobre uma idade em que alguns pacientes não obtêm benefício com a colonoscopia de prevenção: homens ≥85 anos e mulheres ≥ 90 anos.
A colonoscopia de prevenção é potencialmente benéfica em homens ≤85 anos e mulheres ≤ 90 anos, sempre considerando as comorbidades e expectativa de vida, caso não tenha registro de uma colonoscopia sem pólipos nos últimos 10 anos. Pacientes mais jovens têm uma diminuição maior na expectativa de vida do que pacientes idosos após um diagnóstico de câncer colorretal. Ao mesmo tempo, há uma redução de 75% no benefício da triagem para pacientes idosos em comparação com pacientes mais jovens.
Estudos mostraram que o benefício da triagem, após uma colonoscopia sem pólipos inicial, é reduzido em pacientes idosos, pois esse grupo tem maior chance de morrer de outras doenças além do câncer colorretal.
Em resumo: a saúde, expectativa de vida, condição funcional e idade são considerados quando se pensa em interromper o rastreamento do câncer colorretal. Recomenda-se uma abordagem individualizada reservando a colonoscopia a pacientes idosos (≥ 75 anos) saudáveis, quando a expectativa de vida for de 10 anos ou mais.
Quando repetir a colonoscopia no preparo de cólon inadequado?
O preparo inadequado esta associado a perdas de lesões nas colonoscopias. É recomendado repetir entre 6 meses a um anos.
PSOF (pesquisa de sangue oculto nas fezes) positiva antes de completar o intervalo recomendado.
É muito comum em nosso meio. O paciente está em seguimento pós-polipectomia, mas outro médico pede como parte de algum check-up a PSOF. Se a primeira colonoscopia foi ideal este resultado é desconsiderado. Não existem dados que suporte a antecipação da colonoscopia visando o diagnóstico de câncer ou adenoma avançado devido as altas taxas de falso positivo da PSOF.
A colonoscopia só deve ser antecipada se o primeiro exame foi incompleto, teve o preparo de cólon inadequado, existir história familiar de câncer colorretal e pela insegurança de paciente e do médico, principalmente quanto ao local onde a primeira colonoscopia foi realizada. Os pacientes e familiares devem ser alertados a não realizar a PSOF entre as colonoscopias.
Sintomas entre o intervalo de vigilância (sangramento anal, dor abdominal, diarreia e constipação).
Novas doenças podem surgir no intervalo de seguimento. Estes sintomas criam um dilema clínico, mas quando a primeira colonoscopia foi ideal o diagnóstico de câncer ou adenoma avançado da colonoscopia antecipada será provavelmente baixa. A maioria dos médicos opta pela antecipação da colonoscopia devido a possibilidade de novas doenças surgirem.
Outros fatores de risco (uso de AINE ou aspirina, raça, idade e sexo).
Não existe evidência científica que justifique alterar os intervalos recomendados.
Isenção de responsabilidade
As informações contidas neste artigo são apenas para fins educacionais e não devem ser usadas para diagnóstico ou para orientar o tratamento sem o parecer de um profissional de saúde. Qualquer leitor que está preocupado com sua saúde deve entrar em contato com um médico para aconselhamento.